
Abstrak Grafis
Ikatan CH H (HB) biasanya jauh lebih lemah daripada ikatan yang melibatkan gugus OH. Perhitungan kimia kuantum membahas jenis substituen, dan penempatannya, yang dapat menyesuaikan kekuatan HB OH··N dan CH··N agar keduanya hampir setara.

Abstrak
Kemampuan gugus CH untuk bertindak sebagai donor proton kini diterima secara luas, bahkan jika ikatan H (HB), yang dibentuknya biasanya jauh lebih lemah daripada ikatan gugus hidroksil, khususnya untuk C yang terhibridisasi sp 3 . Nukleofil NH 3 dibiarkan mendekati gugus metil terminal dan hidroksil n-butanol, sehingga membentuk CH··N atau OH··N HB. Perhitungan teori fungsi kerapatan menunjukkan bahwa yang terakhir jauh lebih kuat daripada yang pertama. Namun, kekuatan CH··N HB dapat diperkuat dan mendekati lebih dekat dengan OH··N dengan penempatan substituen penarik dan pendonor elektron yang sesuai pada butanol. Energi interaksi CH··N HB mencapai di atas 6–8 kkal mol −1 dalam beberapa kasus, jauh lebih besar daripada prototipe HB dalam dimer air.
1 Pendahuluan
Ikatan hidrogen (HB) tampak penting di banyak subbidang yang meliputi semua kimia dan biologi. Seseorang tidak dapat mulai memahami reaksi apa pun yang terjadi dalam media berair tanpa deskripsi terperinci tentang ikatan H antara molekul air dan reaktan, serta berbagai zat antara di sepanjang profil reaksi. Reaksi tersebut mencakup proses enzim yang sangat penting bagi kehidupan. Struktur yang diadopsi oleh protein, yang menentukan fungsinya, sebagian besar dikendalikan oleh HB antara gugus peptida, atau gugus rantai sampingnya. Integritas dan transmisi kode genetik bertumpu pada HB yang sangat spesifik dalam pasangan basa asam nukleat. Ini, bersama dengan banyak proses penting lainnya di mana HB memainkan peran integral, telah memotivasi sejarah studi yang panjang dan bertingkat yang telah berlangsung lebih dari satu abad. [ 1 – 7 ] Seiring dengan kemajuan penelitian ini, segera disadari bahwa atom A dan D yang terlibat dalam AH··D HB mencakup lebih dari sekadar F, O, dan N yang sangat elektronegatif yang merupakan bagian tak terpisahkan dari formulasi aslinya. Deskripsi modern HB ini mencakup berbagai atom lain di seluruh tabel periodik, seperti S dan Se, semua halogen, dan bahkan atom logam juga. [ 8 – 21 ]
Dari semua pendatang baru di bidang ini, atom C mungkin yang paling menarik dan paling penting. [ 22 – 32 ] Pertama-tama, gugus CH bisa dibilang yang paling umum dalam semua kimia, terutama dalam bidang kimia organik, jadi kemungkinan CH bertindak sebagai donor proton dalam HB akan memiliki implikasi yang luas. Faktor lainnya adalah elektronegativitas atom C dan H yang hampir sama. Fenomena ini akan meminimalkan polaritas dalam ikatan CH, sehingga menetralkan muatan positif parsial pada proton jembatan yang dianggap penting untuk pembentukan HB. Karena alasan inilah gagasan donor proton CH tidak diperhatikan selama bertahun-tahun. Memang, beberapa saran awal untuk efek ini [ 22 , 33 – 41 ] awalnya ditolak mentah-mentah. [ 42 – 44 ] Namun, beberapa dekade terakhir telah menyaksikan kebangkitan kembali konsep ini, lengkap dengan berbagai jenis bukti, yang tidak mungkin diabaikan. Sementara HB yang dibentuk oleh donor proton CH umumnya lebih lemah daripada situasi paralel yang melibatkan OH atau NH, mereka tetap substansial dan dapat memberikan kontribusi besar, terutama jika jumlahnya banyak.
Studi terbaru CH HB telah menghasilkan sejumlah kesimpulan menyeluruh. Pertama-tama, hibridisasi C merupakan faktor penting, dan HB melemah dalam urutan sp > sp 2 > sp 3 . [ 45 ] Seperti donor HB lainnya, penempatan substituen penarik elektron dekat dengan situs CH memperkuat HB yang baru jadi. Salah satu aspek yang lebih menarik dari CH HB adalah kecenderungannya untuk membalikkan karakteristik khas tertentu dari HB secara umum. Artinya, alih-alih pergeseran merah yang diharapkan dari frekuensi peregangan CH, tidak jarang pita ini bergeser ke biru. [ 46 – 55 ] Faktanya, anomali nyata inilah yang menyebabkan beberapa klaim awal [ 56 , 57 ] bahwa interaksi ini sama sekali bukan HB, tetapi lebih baik disebut sebagai “anti-HB”, “tidak tepat”, atau semacamnya. Namun, penyelidikan yang lebih rinci selama bertahun-tahun telah sepenuhnya memverifikasi sifatnya sebagai HB sejati yang semua sifat lainnya sesuai dengan pola yang biasa. [ 58 – 67 ] Singkatnya, arah pergeseran frekuensi peregangan C H, seperti semua HB lainnya, adalah hasil dari dua efek yang saling bersaing, satu mendorong ke arah merah dan yang lainnya ke arah biru. Arah pergeseran yang sebenarnya, untuk semua HB, hanya muncul dari keseimbangan yang rumit antara kedua gaya ini.
Pertanyaan yang masih menggantung dan menonjol menyangkut batas atas kekuatan CH HB. Hidrogen pada C yang dihibridisasi sp 3 menjadi perhatian khusus karena dua alasan. Pertama, hidrogen merupakan gugus CH yang paling umum, yang muncul pada gugus alkil dengan panjang bervariasi dan dalam berbagai situasi. Kedua, hibridisasi ini menghasilkan donor proton CH yang terlemah, sehingga kekuatan potensialnya menjadi dasar bagi beberapa donor yang lebih kuat dengan hibridisasi C lainnya. Pertanyaan yang dibahas di sini adalah apakah gugus CH dapat menggantikan kemampuan donor proton yang biasanya jauh lebih kuat dari gugus OH hidroksil. Apakah ada keadaan di mana CH dapat bersaing dengan sukses melawan OH untuk mendapatkan gugus akseptor proton? Untuk menjawab pertanyaan ini, molekul butanol diambil sebagai prototipe. Perhitungan kimia kuantum secara pasti menunjukkan bahwa OH membentuk HB yang jauh lebih kuat dengan basa daripada gugus metil terminalnya. Kemudian, berbagai substituen ditambahkan di berbagai lokasi dalam molekul, memantau bagaimana gangguan ini memengaruhi persaingan antara CH dan OH untuk mendapatkan basa. Hasilnya menetapkan parameter untuk kondisi yang mungkin diperlukan untuk membalik tatanan stabilitas normal ini dan menetapkan batas atas ikatan CH H tersebut.
2 Bagian Eksperimen
Rangkaian program Gaussian 16 [ 68 ] digunakan untuk melakukan kalkulasi kimia kuantum, menerapkan teori fungsi kerapatan M06-2X [ 69 ] bersama dengan basis set triple-ζ def2-TZVP. M06-2X berulang kali dinilai sebagai salah satu fungsi paling akurat untuk ikatan H dan interaksi nonkovalen terkait. [ 70 – 78 ] Geometri dioptimalkan sepenuhnya dan dikarakterisasi sebagai minima sejati melalui analisis vibrasi harmonik yang menghasilkan semua frekuensi positif. Energi interaksi E int sama dengan selisih antara energi dyad dan jumlah energi kedua monomer dalam geometri yang diadopsi dalam kompleks; E int dikoreksi untuk kesalahan superposisi basis set dengan resep penyeimbang standar. [ 79 ]
Program Multiwfn [ 80 ] menemukan dan mengukur potensi elektrostatik molekular (MEP) maksimum pada permukaan isodensitas ρ = 0,001 au dari setiap monomer yang diisolasi. Lintasan ikatan atom dalam molekul (AIM) dan sifat titik kritis terkait dievaluasi melalui program AIMAll. [ 81 ] Analisis orbital ikatan alami (NBO) dari transfer muatan interorbital [ 82 , 83 ] dilakukan melalui rutinitas NBO yang dimasukkan ke dalam paket Gaussian. Perisai resonansi magnetik nuklir (NMR) dinilai dalam kerangka skema orbital atom invarian pengukur. [ 84 , 85 ]
3 Hasil
3.1 Sifat Monomer
Molekul butanol diilustrasikan dalam Gambar 1 , di mana simbol X menunjukkan tempat substitusi telah dibuat. Substitusi ini terjadi pada dua situs C yang melekat pada OH, dan pada situs-situs pada atom karbon CH. Substituen dipilih dengan anggapan bahwa gugus penarik elektron (EWG) pada C tertentu akan menarik kerapatan elektron dari CH atau OH di dekatnya, dengan demikian meningkatkan muatan positif pada H sehingga menjadikannya donor proton yang lebih baik. Daftar substituen ini ditampilkan dalam dua kolom pertama Tabel 1 di mana X CH dan X OH menunjukkan pasangan situs mana yang sedang ditempati. Substitusi ini dibuat berpasangan di mana misalnya CN yang tercantum untuk X CH di baris kedua menempatkan sepasang gugus siano pada C yang bersangkutan.

Gambar 1
Buka di penampil gambar
Presentasi PowerPoint
Diagram molekul n-butanol, dengan situs pelabelan X untuk substitusi.
Tabel 1. Maksimum MEP pada proton yang ditunjukkan, jarak intermolekul R(H··N) dalam kompleks dengan NH 3 , dan energi interaksi dalam dyad.
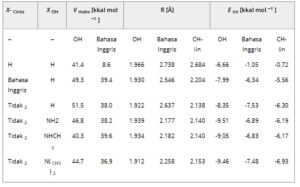
Salah satu sifat yang membuat proton tertentu efektif sebagai jembatan dalam HB adalah MEP positif yang mengelilinginya dalam molekul asam. Sifat ini dikuantifikasi sebagai Vmax , maksimum MEP pada permukaan isodensitas 0,001 au. Seperti terlihat dari baris pertama Tabel 1 , Vmax jauh lebih besar pada hidroksil H daripada pada metil H, 41,4 versus 8,6 kkal mol −1 . Memindai entri dalam Tabel 1 menunjukkan bahwa hidroksil Vmax ditingkatkan oleh EWG bahkan ketika mereka berada di posisi X CH yang lebih jauh . Misalnya , substituen NO2 meningkatkan Vmax sebesar 10 kkal mol −1 . Karena jaraknya yang lebih dekat, EWG yang sama ini memiliki efek yang jauh lebih kuat pada V maks metil H, meningkatkan kuantitas ini hingga 30 kkal mol −1 . Namun meskipun begitu, MEP proton hidroksil tetap jauh lebih tinggi, 51,5 dibandingkan dengan 38,0 kkal mol −1 .
Dengan cara yang analog, penempatan gugus pendonor elektron (EDG) seharusnya mengurangi potensial positif pada proton-proton ini. Dengan mempertahankan gugus NO2 pada posisi X CH , EDG yang semakin kuat ditambahkan ke situs X OH . Karena atom-atom H bermutasi ke berbagai gugus amina, Vmax proton hidroksil diturunkan ke 40 detik. Karena situs X OH ini jauh dari metil, MEP pada proton CH tidak terlalu terpengaruh, bahkan sedikit membesar untuk NHCH3 . Namun yang penting, bahkan dengan penempatan EWG yang kuat di dekat metil, dikombinasikan dengan EDG di dekat OH, Vmax proton hidroksil tetap jauh lebih tinggi daripada CH, bahkan jika perbedaan antara keduanya dapat diturunkan hingga sekecil 0,7 kkal mol −1 .
3.2 Geometri dan Energi Dyad
Ketika NH3 diizinkan untuk berinteraksi dengan situs OH dan CH, geometri yang dioptimalkan dari dyad yang dihasilkan ditampilkan dalam Gambar 2 dan 3. Jarak HB, yang didefinisikan sebagai R(H··N), dirangkum dalam dua kolom berikutnya dari Tabel 1 , diikuti oleh energi interaksi. HB ke gugus hidroksil secara universal lebih pendek daripada jarak CH··N, dalam jumlah antara 0,25 dan 0,77 Å. Demikian pula, HB OH··N lebih kuat daripada rekan CH··N mereka. Dalam butanol yang tidak tersubstitusi, energi interaksi ini masing-masing adalah 6,7 dan 1,1 kkal mol −1 . Penambahan berbagai substituen meningkatkan energi H··N ini, terutama dengan interaksi CH··N yang menunjukkan peningkatan yang sangat penting, mencapai puncak 7,5 kkal mol −1 . Memang energi interaksi CH··N untuk semua butanol tersubstitusi setidaknya 6,3 kkal mol −1 . Jadi tampaknya meskipun CH··N HB ini tetap lebih lemah daripada ikatan OH··N untuk molekul apa pun, mereka masih dapat mencapai besaran yang cukup luar biasa untuk gugus CH hibridisasi sp 3 . Ide ini konsisten dengan serangkaian perhitungan baru-baru ini [ 27 ] mengenai CH··N HB dalam F 3 CH··NCLi.

Gambar 2
Buka di penampil gambar
Presentasi PowerPoint
Geometri kompleks NH 3 yang dioptimalkan dengan a,d) n-butanol, b,e) gugus CN di situs X CH , dan c,f) gugus NO 2 di situs X CH . Jarak dalam Å dan sudut dalam derajat

Gambar 3
Buka di penampil gambar
Presentasi PowerPoint
Geometri kompleks NH 3 yang dioptimalkan dengan a,d) gugus NO 2 pada situs X CH dan NH 2 pada situs X OH , b,e) gugus NO 2 pada situs X CH dan NHCH 3 pada situs X OH , dan c,f) gugus NO 2 pada situs X CH dan N(CH 3 ) 2 pada situs X OH . Jarak dalam Å dan sudut dalam derajat.
Harus dipahami bahwa meskipun NH3 adalah molekul kecil, tidak rentan untuk terlibat dalam interaksi sekunder, yang terakhir tidak dapat sepenuhnya dihindari dan dapat berkontribusi pada energi interaksi dari beberapa diade ini. Gambar 2 dan 3 menempatkan garis biru putus-putus untuk semua kontak antarmolekul yang AIM mengidentifikasi jalur ikatannya. Jelas dari sisi kiri Gambar 2 bahwa satu-satunya jalur ikatan antarmolekul untuk OH··N HB sebenarnya adalah ikatan OH··N itu sendiri. Namun, hal yang sama tidak dapat dikatakan untuk kontak CH··N di sisi kanan Gambar 2. Misalnya, ada NH··C HB pada Gambar 2d, ikatan tetrel N··C pada Gambar 2e , dan ikatan pnikogen N··N pada Gambar 2f . Kepadatan titik kritis ikatan (BCP) dirangkum dalam Tabel 2 untuk HB ini. Bagi sistem yang berisi jalur ikatan AIM sekunder, jumlahnya tercantum dalam tanda kurung. Kuantitas yang tercantum dalam Tabel 2 menunjukkan bahwa interaksi ikatan sekunder ini tidak dapat sepenuhnya diabaikan dalam menganalisis asal energi interaksi total. Setelah menambahkan gugus amina ke situs XOH, interaksi sekunder ini mulai memainkan peran dalam energi OH··N HB, seperti yang terlihat di sisi kiri Gambar 3 , terutama dalam bentuk NH··N HB. Interaksi sekunder utama dalam kasus CH··N di sisi kanan Gambar 3 melibatkan ikatan pnikogen N··O ke atom O dari gugus nitro, meskipun ada juga beberapa CH··N HB tambahan pada Gambar 3f .
Tabel 2. Kepadatan titik kritis ikatan utama untuk dyad. Jumlah kepadatan BCP sekunder dalam tanda kurung. Nilai NBO dari energi pemindahan muatan orde kedua dari N pasangan elektron bebas NH 3 ke orbital σ*(OH/CH).

Secara keseluruhan, pola kerapatan AIM pada Tabel 2 cukup sesuai dengan energetika. Kerapatan bervariasi antara 0,028 dan 0,032 au untuk HB OH··N dan cenderung meningkat saat E int menjadi lebih negatif. Kerapatan CH··N secara seragam lebih kecil daripada OH··N, konsisten dengan energi interaksinya yang lebih kecil. Ada lonjakan yang cukup besar pada ρ (CH··N) setelah penambahan gugus amino pada posisi X OH , yang tidak bertepatan dengan lonjakan E int tersebut .
Tiga kolom terakhir dari Tabel 2 berisi energi gangguan orde kedua yang dihitung oleh NBO untuk transfer muatan dari pasangan bebas amonia N ke orbital antiikatan σ*(OH/CH) dari butanol tersubstitusi. Nilai-nilai ini cukup besar untuk OH HB, antara 14 dan 18 kkal mol −1 , dan mencerminkan kenaikan energi interaksi yang ditunjukkan pada Tabel 1 . Kuantitas ini jauh lebih kecil untuk CH HB, bahkan dalam kasus di mana energi interaksi kompetitif dengan OH··N. Mengenai kerapatan AIM, ada lompatan kuantum dalam E2 ketika gugus amina ditambahkan ke situs X OH .
Jika digabungkan, data AIM dan NBO memperkuat gagasan bahwa OH··N HB tetap lebih kuat daripada CH··N, meskipun terdapat substituen EWG atau EDG pada molekul tersebut. Perbedaan ini secara khusus ditegaskan oleh perlakuan NBO yang menandakan margin lebar antara kedua jenis ikatan tersebut.
3.3 Pengukuran Kekuatan Ikatan Secara Spektroskopi
Pembentukan HB diketahui dapat menyebabkan gangguan tertentu pada molekul donor proton. Yang paling menonjol adalah peregangan ikatan kovalen OH, dan pergeseran merah terkait pada frekuensi peregangannya. Pola untuk ikatan CH tidak konsisten, karena ikatan ini sering mengalami perubahan ke arah yang berlawanan, yaitu kontraksi dan pergeseran biru. Perilaku yang berlawanan ini paling sering dikaitkan dengan hibridisasi sp3 atom C , seperti yang terjadi di sini.
Perubahan dalam dua parameter kunci ini disorot dalam Tabel 3 untuk dua jenis sistem HB. Perilaku ikatan OH cukup seperti yang diharapkan. Ikatan ini meregang dalam jumlah antara 0,012 dan 0,015 Å, dan peregangan ini cenderung meningkat seiring dengan parameter energik dan parameter lain yang mengukur kekuatan HB. Hal yang sama dapat dikatakan untuk pergeseran merahnya yang konsisten, yang bervariasi antara −244 dan −332 cm −1 .
Tabel 3. Perubahan panjang ikatan OH dan CH, dan frekuensi peregangannya, yang disebabkan oleh kompleksasi dengan NH 3 .

Perilaku ikatan CH sangat berbeda. Tidak seperti OH, ikatan CH mengalami kontraksi kecil untuk tiga sistem di bagian atas Tabel 3 , sebanyak 0,002 Å. Perubahan ini disertai dengan pergeseran biru frekuensi peregangannya, antara 8 dan 32 cm −1 . Namun, arah perubahan terbalik untuk tiga baris terakhir Tabel 3 di mana ikatan CH meregang dan pergeseran merah yang signifikan terjadi pada frekuensi terkaitnya. Perlu dicatat bahwa transisi ini merupakan karakteristik dari keberadaan substituen amina, yang cocok dengan penanda AIM dan NBO dari ikatan CH··N yang ditingkatkan yang diuraikan sebelumnya.
Pengukur spektroskopik lain dari kekuatan HB berasal dari data NMR. Secara umum diterima bahwa proton penjembatan mengalami pergeseran medan bawah yang disebabkan oleh pembentukan HB, dan bahwa pelepasan perisai ini secara kasar proporsional dengan kekuatan ikatan. Ada penelitian lain yang telah menunjukkan bahwa atom N penerima proton juga mengalami pelepasan perisai, sekali lagi terkait erat dengan kekuatan ikatan. [ 86 – 89 ] Perubahan dalam perisai kimia yang timbul sebagai akibat dari kompleksasi ini dikompilasi dalam Tabel 4 untuk proton penjembatan dan atom N amonia. Konsisten dengan ukuran kekuatan HB lainnya yang dibahas sebelumnya, pelepasan perisai proton OH secara substansial lebih besar daripada untuk CH. Pelepasan perisai OH terletak pada kisaran 5–6 ppm, sedangkan CH adalah 3 ppm atau kurang. Pada catatan yang sama, pergeseran medan bawah dari N penerima proton juga lebih besar dalam kasus HB OH··N. Deshielding ini berkisar hingga 20 ppm untuk OH dan berada dalam kisaran 4–13 ppm untuk CH.
Tabel 4. Perubahan perisai kimia NMR (ppm) setelah kompleksasi dengan NH 3 .

4 Diskusi
Pemeriksaan geometri diadik CH··N pada Gambar 2 dan 3 segera memperjelas bahwa ikatan ini jauh dari linear. Sudut CH··N bervariasi antara 104° dan 150°. Beberapa distorsi dari linearitas ini tidak diragukan lagi disebabkan oleh kemungkinan interaksi sekunder yang terwujud dalam diagram AIM dan dikuantifikasi dalam Tabel 2. Interaksi tambahan ini juga cenderung menggeser N dari ikatan CH dan memutar pasangan bebas N menjauh dari CH. Dalam upaya untuk mendapatkan pemahaman yang lebih jelas tentang CH··N HB sebagai entitas murni, serangkaian optimasi geometri dilakukan dengan pembatasan HB linear; yaitu, N dibatasi untuk terletak di sepanjang sumbu ikatan C H.
Hasil-hasil ini tercantum dalam kolom berlabel CH-lin pada Tabel 1–4 . Seperti yang jelas dari Tabel 1 , linearisasi paksa ini pertama-tama mengurangi panjang HB, beberapa di antaranya cukup substansial. Efek lain yang lebih penting adalah penurunan energi interaksi, tetapi hanya dalam jumlah kecil. Dengan kata lain, hilangnya interaksi sekunder sebagian besar dikompensasi oleh penguatan CH··N. Penguatan terakhir ini jelas dari kerapatan BCP dan kuantitas E2 pada Tabel 2. Efek ini sangat dramatis untuk tiga baris pertama tabel tempat situs X OH ditempati oleh H. Peralihan dari susunan CH··N yang bengkok ke linear juga menghasilkan temuan bahwa semua ikatan mengalami pergeseran merah: Semua ikatan CH meregang dan mengalami pengurangan dalam frekuensi peregangan CH. Efek-efek ini tetap lebih kecil daripada yang ditemui pada HB OH··N tetapi tetap signifikan. Dan akhirnya, linearitas paksa HB menyebabkan pergeseran medan proton yang lebih besar, seperti terlihat pada Tabel 4 , lagi-lagi terutama terlihat ketika tidak ada substitusi pada atom karbon OH.
Singkatnya, seseorang dapat mempertimbangkan CH··N HB ini sebagai gaya stabilisasi sejati, dengan energi interaksi melebihi 5 kkal mol −1 , setidaknya untuk yang mengandung EWG di situs X CH . Ketika dalam susunan linier, ikatan ini menampilkan peregangan dan pergeseran merah ikatan kovalen CH, seperti halnya OH··N HB konvensional, dan pelepasan proton penghubung berada pada orde 3 ppm. Namun, ikatan CH··N ini tidak cukup terarah untuk mempertahankan linearitasnya. Ketersediaan ikatan nonkovalen sekunder, bahkan yang lemah, mendorongnya menjauh dari linearitas, sehingga mengurangi beberapa gejala spektroskopi dan gejala lain dari keberadaannya. Meskipun demikian, CH··N HB inilah yang mewakili bagian utama dari total energi ikat.
5 Kesimpulan
Tidak seperti hidroksilnya, gugus CH dari butanol adalah donor proton yang buruk. Energi interaksinya dengan NH3 hanya 1,0 kkal mol −1 . Kepadatan AIM dari CH··N adalah 0,0074 au dan E2 untuk transfer muatan hanya 0,6 kkal mol −1 . Ikatan C H mengalami kontraksi kecil dan pergeseran biru sekitar 10 cm −1 . Sebaliknya, ikatan ini diperkuat secara nyata oleh penempatan EWG pada C. Energi HB-nya naik hingga 7,5 kkal mol −1 , dalam 1 kkal mol −1 dari energi interaksi OH··N dari asam yang sama. Namun, HB ini sangat tertekuk, sehingga pergeseran medan bawah sinyal NMR proton CH agak kecil pada hanya 1 ppm atau kurang, dan ikatan CH mengalami kontraksi dan pergeseran biru. Masalah terakhir dapat diatasi jika CH··N berada dalam penyelarasan linier, yang membuat CH··N lebih mirip perilakunya dengan OH··N, dengan pergeseran merah dan pergeseran medan bawah protonnya sekitar 3 ppm.
Penambahan EDG di ujung lain molekul tidak banyak mengubah energi CH··N HB, tetapi memperkuat penanda non-energi. Energi transfer muatan NBO meningkat secara dramatis, dan ada peningkatan substansial dalam kerapatan titik kritis ikatan CH··N, keduanya dihasilkan dari linearitas dekat CH··N HB ini. Penyelarasan yang ditingkatkan ini juga membalikkan respons ikatan kovalen CH, mengubah kontraksi ikatan menjadi peregangan, dan mengubah pergeseran biru menjadi pergeseran merah. Meskipun CH··N HB tetap lebih lemah daripada OH··N untuk semua sistem yang diperiksa, mereka dapat menjadi sangat kuat dengan substitusi yang tepat, hingga sebanyak 7,5 kkal mol −1 , dan dapat mendekati kekuatan ikatan OH··N dalam waktu kurang dari 1 kkal mol −1 .

